 From
From Andrew L. Mammen, MD, PhD Assistant Professor of Neurology and Medicine; Co-Director; Johns Hopkins Myositis Center, Baltimore, Maryland
06/08/2011;Abstract
The different autoimmune myopathies—for example, dermatomyositis, polymyositis, and immune-mediated necrotizing myopathies (IMNM)—have unique muscle biopsy findings, but they also share specific clinical features, such as proximal muscle weakness and elevated serum levels of muscle enzymes.Furthermore, around 60% of patients with autoimmune myopathy have been shown to have a myositis-specific autoantibody, each of which is associated with a distinct clinical phenotype.
The typical clinical presentations of the autoimmune myopathies are reviewed here, and the different myositis-specific autoantibodies, including the anti-synthetase antibodies, dermatomyositis-associated antibodies, and IMNM-associated antibodies, are discussed in detail.
This Review also focuses on a newly recognized form of IMNM that is associated with statin use and the production of autoantibodies that recognize 3-hydroxy-3-methylglutaryl-coenzyme A reductase, the pharmacological target of statins.
The contribution of interferon signaling to the development of dermatomyositis and the potential link between malignancies and the initiation of autoimmune myopathies are also assessed.
Introduction
Autoimmune myopathies are a heterogeneous group of diseases, of which polymyositis and dermatomyositis are probably the best known.These two entities share several clinical features, such as proximal muscle weakness that typically progresses over a period of weeks to months, and evidence of inflammation on muscle biopsy.
Immune-mediated necrotizing myopathies (IMNMs) probably represent a distinct form of autoimmune myopathy that is not associated with the same levels of inflammatory infiltrates as polymyositis or dermatomyositis on muscle biopsy.
Inclusion body myositis (IBM) is also a disorder considered by some authors to be a member of this group of diseases.Indeed, IBM muscle biopsies reveal inflammatory infiltrates similar to those found in poly-myositis, and patients with IBM have other evidence of immune system activation.
Nevertheless, patients with IBM, unlike those with dermatomyositis, polymyositis or IMNM, have a unique pattern of weakness and lack a sustained response to immunosuppression, which is the treatment of choice for patients with these conditions.Furthermore, pathological evidence suggests that IBM might actually be a myodegenerative disease that is associated with abnormal accumulation of amyloid-β and/or TAR DNA-binding protein 43, as seen in Alzheimer disease and amyotrophic lateral sclerosis, respectively.
Given that the primary role of the inflammatory response in IBM is currently under debate, this Review focuses primarily on the clinical presentation and pathogenesis of adult-onset polymyositis, dermatomyositis, and IMNM.
The association between distinct clinical phenotypes and autoantibodies is also reviewed.
Furthermore, evidence that statins may trigger a unique form of autoimmune muscle disease is discussed, along with data highlighting the involvement of interferon (IFN) signaling and malignancies in the initiation and maintenance of specific autoimmune myopathies.
Clinical Presentation
In 1975 and 1977, Bohan and Peter published a series of papers that established diagnostic criteria for dermatomyositis and polymyositis ( Box 1 ).Although these criteria are imperfect, they are still widely used in both clinical and research settings, and provide a useful starting point for discussing the typical clinical features associated with autoimmune myopathy.Proximal Muscle Weakness
The most common clinical feature associated with autoimmune myopathies is symmetrical proximal muscle weakness that progresses over a time period of weeks to months.A patient with such a disease may complain that they have difficulty rising from chairs, climbing stairs, or washing their hair. In severe cases of autoimmune myopathy, oropharyngeal weakness can result in dysphagia and/or dysphonia; in such cases diaphragmatic weakness may also occur and require mechanical ventilation.
Although autoimmune myopathies are frequently characterized as 'painless weakness', some patients do have considerable myalgia; thus, the presence of muscle pain should not preclude a diagnosis of autoimmune myopathy.By contrast, muscle weakness slowly evolving over years, asymmetric or distal muscle weakness, facial weakness or scapular winging are rarely associated with autoimmune myopathies and should strongly suggest the possibility of an alternative diagnosis such as limb-girdle muscular dystrophy, or other non-immune-mediated muscle disease.
Electromyography
In patients with dermatomyositis or polymyositis, electromyography (EMG) of the affected muscle typically reveals short-duration, small-amplitude, polyphasic motor units. These motor units are also evident in other myopathic processes, including muscle disuse. In addition, patients with active autoimmune myopathy usually have features on EMG associated with 'irritable myopathy', such as spontaneous activity (fibrillation potentials and positive sharp waves) and/or complex repetitive discharges.Of note, in the experience of this reviewer, patients with partially treated dermatomyositis, polymyositis or steroid myopathy may have a non-irritable myopathy that lacks spontaneous activity on EMG.Muscle Biopsy
In patients with suspected autoimmune muscle disease, a muscle biopsy can provide valuable diagnostic information. Muscle biopsy findings that were recognized by Bohan and Peter to be associated with autoimmune myopathies include: degenerating and/or necrotic myofibers, regenerating muscle fibers, atrophic muscle cells, and evidence of inflammatory exudates.These features are not, however, specific for immune-mediated myopathies, as they can also be found in patients with IBM and inflammatory muscular dystrophies, such as limb-girdle muscular dystrophy type 2B (also called dysferlinopathy).Since Bohan and Peter published their classification scheme, biopsies from patients with dermatomyositis, polymyositis and IMNM have been shown to have unique pathological features, indicating that different pathophysiological mechanisms underlie these distinct diseases. As discussed in detail below, evidence of atrophic, degenerating or regenerating fibers within the perifascicular area is pathognomonic for dermatomyositis (Figure 1).By contrast, muscle biopsies from patients with polymyositis are characterized by the presence of cytotoxic T cells surrounding and invading non-necrotic myofibers (Figure 1b).
Muscle biopsies from patients with IMNM typically show marked myofiber necrosis, degeneration and regeneration, and few, if any, inflammatory cells are usually seen in muscle biopsies from these patients (Figure 1d).
Since toxic myopathies, endocrine-associated myopathies, paraneoplastic myopathies, and muscular dystrophies can also be associated with necrotic myofibers on muscle biopsy, the presence of myositis-specific autoantibodies (MSAs, see below) can help differentiate between these diseases and disorders that have an immune-mediated pathology.
When specific features of dermatomyositis, polymyositis or IMNM are absent, non-immune-mediated muscle diseases should always be considered as an alternative diagnosis. Moreover, features on muscle biopsies that are clearly characteristic of other muscle diseases, such as rimmed-vacuoles (as seen in IBM) or increased accumulation of glycogen (as seen in acid maltase deficiency), should suggest that the patient does not have dermatomyositis, polymyositis or IMNM.
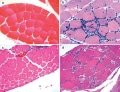
| Figure 1. Muscle Biopsies From Patients With Polymyositis, Dermatomyositis or Immune-mediated Necrotic Myopathy. a | A typical muscle fascicle from a normal muscle biopsy specimen includes myofibers of uniform size. b | The presence of lymphocytes (the small blue cells in this hematoxylin and eosin stain) surrounding and invading muscle fibers is a characteristic feature of polymyositis muscle biopsies, whereas c | perifascicular atrophy is typically seen in muscle biopsies from patients with dermatomyositis. d | Degenerating, necrotic and regenerating muscle fibers are a characteristic feature of muscle biopsies from patients with immune-mediated necrotic myopathy. |
Elevated Muscle Enzymes
Elevated serum levels of muscle enzymes such as creatine kinase, aldolase, aspartate transaminase and/or alanine transaminase are present in at least 90% of patients with autoimmune myopathy. Although elevated levels of creatine kinase are believed to be the most sensitive and specific marker of muscle damage, patients with autoimmune myopathies can present with elevated serum aldolase levels without an accompanying increase in serum creatine kinase levels.In 2009, a series of 12 patients with elevated serum aldolase levels but normal levels of creatine kinase was studied in detail.This analysis showed that many of these patients had muscle pain (92%) as well as arthralgias (75%) and interstitial lung disease (42%); only 50% of the patients had muscle weakness on examination.
Muscle biopsies showed that fragmentation of perimysial connective tissue and elevated acid phosphatase cellularity was prominent in those patients with selectively high serum aldolase levels. Furthermore, a few of the patients with high levels of serum aldolase were shown to have perifascicular atrophy on muscle biopsy, or skin rashes suggestive of dermatomyositis.
Importantly, all patients responded to corticosteroid therapy. Taken together, these findings indicate that in patients with muscle discomfort and normal serum levels of creatine kinase, measuring serum aldolase levels might help identify patients with a steroid-responsive autoimmune myopathy.
In patients with autoimmune myopathy, identifying whether elevated serum levels of transaminases are the result of muscle or liver disease can be challenging, particularly in those patients taking potentially hepatotoxic medications, such as methotrexate or azathioprine. However, as the liver enzyme γ-glutamyl transpeptidase (GGT) is not released by damaged muscle fibers,elevated serum levels of GGT should suggest the possibility of concurrent liver damage.
MRI
Although not included in the Bohan and Peter criteria, MRI might help identify and, thus, aid the management of patients with autoimmune myopathy. On short tau inversion recovery (STIR) imaging, increased signal intensity within muscle tissue is consistent with the presence of muscle necrosis, degeneration, and/or inflammation (Figure 2).As a result, this finding has been incorporated into contemporary diagnostic criteria for autoimmune myopathies.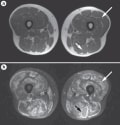
| Figure 2. Thigh MRI From a Patient With Dermatomyositis. a | In T1-weighted images, fat is bright and muscle is dark. b | In short tau inversion recovery sequences, normal muscle is dark and inflamed muscle is bright. Long arrows indicate the inflamed left vastus lateralis muscle. Short arrows highlight the left biceps femoris muscle; the bright rim around this muscle is consistent with fascial inflammation. |
A study of patients with autoimmune myopathy (polymyositis or dermatomyositis) has revealed that inflammatory cells are abundant in areas with high-intensity STIR signal on MRI; however, inflammatory cells, albeit fewer in number, were also shown to be present in areas deemed not to be affected on MRI. The same investigators also found that MRI signal intensity decreased in patients with dermatomyositis or polymyositis after treatment had been initiated;this finding suggests that MRI could help the treating clinician to assess a clinical response. Nevertheless, further studies are required before the utility of MRI in informing decision-making relating to the treatment of autoimmune myopathies is known.
Dermatomyositis Rash
Characteristic cutaneous features often help the treating clinician to differentiate patients with dermatomyositis from those with polymyositis or IMNM.Purplish discoloration around the eyes known as a heliotrope rash and/or an erythematous rash over the extensor surfaces of the metacarpophalangeal, proximal interphalangeal and distal interphalangeal joints referred to as Gottron papules (Figure 3) are both features of dermatomyositis. In fact, the heliotrope rash and Gottron papules are the only two cutaneous findings that are specific for this disorder. Indeed, patients with these features who lack muscle involvement are referred to as having 'amyopathic' dermatomyositis.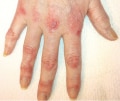
| Figure 3. Gottron Papules. In a patient with dermatomyositis, Gottron papules—pathognomonic cutaneous manifestations of dermatomyositis—are evident on the extensor surfaces of the metacarpophalangeal, proximal interphalangeal and distal interphalangeal joints. |
Overlap Syndromes
Patients with autoimmune myopathy can present with, or develop, an overlap autoimmune rheumatic disease such as scleroderma, systemic lupus erythematosus, Sjögren syndrome, rheumatoid arthritis, or mixed connective disease.Thus, patients with autoimmune myopathy can have typical features of the coexisting rheumatic disease; for example, dryness of the eyes and mouth (Sjögren syndrome) or kidney involvement (systemic lupus erythematosus), as well as symptoms associated with immune-mediated muscle disease.
The frequency with which autoimmune muscle disease occurs in the context of other rheumatic diseases has not been well-defined. For example, in the case of scleroderma, skeletal muscle involvement has been reported to occur in 16-93% of patients, depending on the diagnostic criteria used to classify the condition.
Patients with autoimmune myopathies, especially those with dermatomyositis or polymyositis, might also present with cardiological symptoms including conduction defects, arrhythmias, and reduced ejection fractions Furthermore, interstitial lung disease (ILD) occurs in a substantial number of patients with dermatomyositis or polymyositis. This condition is typically thought to occur in 5-46% of patients with either of these conditions,and the incidence of pulmonary symptoms seems to depend on the clinical setting and the criteria used to determine pulmonary involvement. In one study, when abnormalities on high-resolution CT and/or pulmonary function tests were used to diagnose ILD (even in the absence of symptoms), 11 of 17 (65%) patients with dermatomyositis or polymyositis were shown to have ILD.Importantly, ILD most often occurs in the context of anti-Jo-1 or one of the other antisynthetase autoantibodies (see below).
Autoantibodies
Since Bohan and Peter's diagnostic criteria for dermatomyositis and polymyositis were developed, it has become clear that patients with autoimmune myopathies frequently have autoantibodies. Myositis-associated auto-antibodies (for example, anti-Ro and anti-La) are associated with both immune-mediated myopathies and other connective tissue disorders and will not be discussed further here. By contrast, each MSA is associated with a unique clinical phenotype, and these autoantibodies are found almost exclusively in patients with immune-mediated myopathy or antisynthetase syndrome. New autoantibodies are continually being identified and, to date, around 60-80% of patients with autoimmune myopathy seem to have at least one MSA.In fact, several classification schemes have proposed that the presence of MSAs be included in inclusion criteria for dermatomyositis and polymyositis.Interestingly, with the exception of anti-155/140, MSAs are associated with a decreased risk of malignancy.The individual MSAs are discussed in detail below.Statin-associated IMNM
Musculoskeletal symptoms such as myalgia and cramp are quite common in patients taking statins (9-20%), but they are usually mild.By contrast, rhabdomyolysis is a well-known severe adverse event associated with statin use. Fortunately, however, this adverse event occurs rarely in patients taking this medication, at a rate of around 0.4 per 10,000 patient years.
In most cases, statin-associated muscle complaints improve when the treatment is discontinued, and complete recovery can be expected within a few weeks or months after discontinuation of the drug.
Nevertheless, over the past two decades, numerous case reports have indicated that statins might cause dermatomyositis or polymyositis in some patients, and the identification of inflammatory cells in muscle biopsies taken from these patients supports this hypothesis.
Several compelling studies have also shown that patients can develop IMNMs after taking statins.
In one case series, eight patients were shown to develop myopathy while taking statins, and in some cases the myopathy persisted or even progressed despite discontinuation of the medication.
In fact, seven of the eight patients only improved on initiation of immunosuppressive therapy. Seven of the eight cases had numerous necrotic and regenerating fibers on muscle biopsy, indicating that they might have had statin-associated IMNM. In five cases, marked inflammation on muscle biopsy was absent, indicating that these patients did not have clinical features associated with dermatomyositis or polymyositis. In all eight cases, major histocompatibility complex class I (MHC-I) expression was present in non-necrotic muscle fibers; this finding is a characteristic feature of immune-mediated muscle diseases, and is not seen in patients with other forms of muscle disease such as the muscular dystrophies
In a second case series, 24 patients were identified who had progressive proximal muscle weakness after starting statins, which progressed even after these medications were discontinued.These patients had elevated serum creatine kinase levels, and muscle biopsies revealed marked myofiber necrosis and regeneration in the absence of prominent lymphocytic infiltrates, consistent with a necrotizing myopathy. The patients' symptoms improved with immunosuppressive medications; however, over 50% of the patients worsened when immunosuppressive therapy was tapered. This series also demonstrated that the prevalence of statin exposure was markedly higher in patients with IMNM than in control patients with dermatomyositis, polymyositis or IBM.
Taking a different approach, researchers at Johns Hopkins University (including myself) have identified novel autoantibodies that recognize 200 kDa and 100 kDa proteins in 16 of 26 patients who presented to our department with a necrotizing myopathy. No other autoantibodies or alternative diagnoses were identified in these patients.The patients who expressed these novel antibodies had proximal muscle weakness and high serum levels of creatine kinase, and responded to immunosuppressive therapy—clinical symptoms worsened in many of the patients when immunosuppressive treatment was tapered. Analysis of muscle biopsies taken from these patients revealed that 75% of the cases had abnormal capillary morphologies, 50% had evidence of MAC deposition on non-necrotic muscle cells, and 50% had MHC-I expression in non-necrotic myofibers.Of note, 63% of the patients who had these novel antibodies had been exposed to statins before developing myopathy. Furthermore, compared with age-matched control patients with myopathy, the prevalence of statin use in patients with anti-200/100 autoantibodies (83%) was significantly higher than in patients with dermatomyositis (25%), polymyositis (37%) or IBM (34%).
In a follow-up study, we identified the autoantigen recognized by the anti-200/100 autoantibody as 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR)—the pharmacological target of statins.
Statins are known to dramatically upregulate HMGCR protein levels; thus, in some patients, increased HMGCR expression could trigger anti-HMGCR autoimmunity. Why some statin-naive patients with IMNM also develop anti-HMGCR autoantibodies remains to be determined.
Of note, elevated HMGCR expression is required for muscle differentiation in vitro.We have shown that in muscle biopsy specimens, regenerating human muscle fibers also express high levels of HMGCR.This finding suggests that after statin medications are discontinued, high levels of HMGCR expression in regenerating muscle tissue might continue to drive the autoimmune response.
Together, the studies mentioned above strongly suggest that an environmental factor—statin medication—is associated with a distinct form of autoimmune necrotizing myopathy that is characterized by the production of anti-HMGCR autoantibodies.
Since associations between environmental factors and the development of sustained autoimmunity are rare, this distinct form of autoimmune necrotizing myopathy might prove to be a model system for studying this phenomenon.
for rest of article : go to:-
http://www.medscape.org/viewarticle/743647_4
1 comment:
Inclusion body myositis treatment on time is important for your muscles. Natural Herbs Clinic paid own duty in the Inclusion Body Myositis Natural Treatment technique with herbal extract without any side effect and extended run safe.
Post a Comment